
Author: Zach Lewis, Director, Regulatory and Compliance
In today’s race to bring advanced therapies to patients, one hurdle continues to plague the industry: Chemistry, Manufacturing, and Controls (CMC).
Among the CRLs FDA published from 2020 to 2024, 74% of published FDA Complete Response Letters (CRLs) cited quality/manufacturing (CMC) deficiencies including process gaps and inspection deficiencies. This made CMC a leading driver of regulatory delays and rejections, often stalling promising therapies at the finish line. This summer, Ultragenyx received a CRL citing CMC and facility observations, and Capricor received a CRL that required additional clinical data and noted outstanding CMC items, despite apparent alignment earlier in review.
As the industry progresses, the FDA is reinforcing the standard that no matter how promising the science, therapies will not be approved commercially without a robust and repeatable manufacturing process. Facility readiness is a recurring trigger, with CRLs frequently citing unresolved observations from pre-approval inspections (PAIs) and GMP inspections. In today’s landscape, approval hinges on operational execution as well as clinical data.
Early Decisions Shape Commercial Success
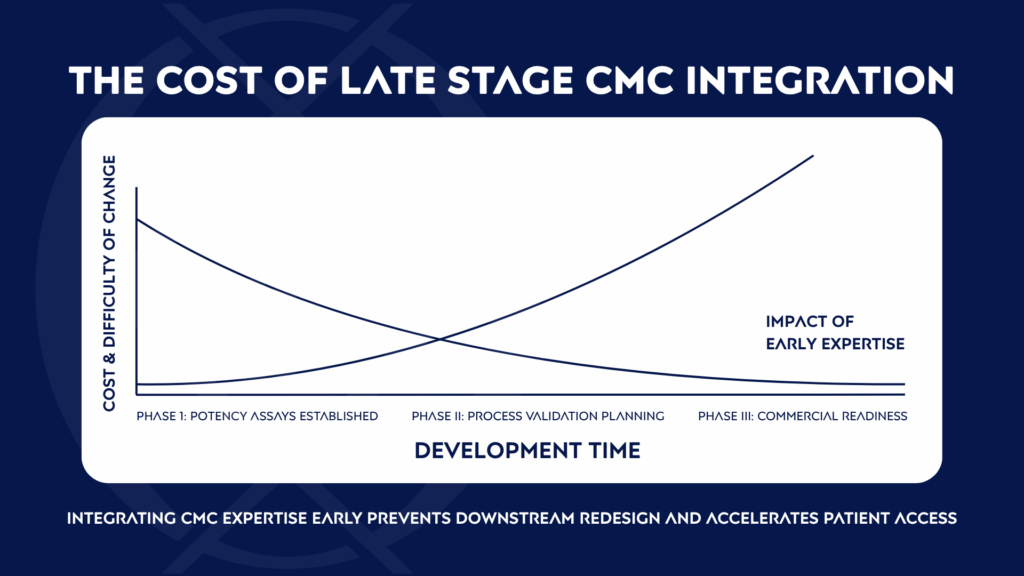
Cell and gene therapy developers often accelerate toward clinical milestones under compressed timelines. It’s understandable, patients are waiting. From the start, developers must be thoughtful about technology selection and whether a process can truly scale commercially. Early design decisions like vector, host cell line, and expansion methods have consequences for scalability, variability, and regulatory success, just as a process that works in a small, highly trained lab team may not translate to GMP manufacturing. CMC rigor cannot be regarded as a late-stage detail, it must be embedded across the product lifecycle.
If those decisions are made without foresight, the result is often a process that looks good in a lab but falters in GMP conditions, where therapies must perform consistently with far less reliance on highly trained scientists at every step. The FDA sees this, and it’s why so many promising applications are delayed.

The Process is the Product: The Discipline of Process Characterization
FDA is increasingly rejecting submissions where the commercial manufacturing process is not well characterized, even when the early clinical data look strong.
Strong CMC execution isn’t about characterizing everything; it’s about characterizing the right things. Developers must design thoughtful experiments that define critical process parameters and set acceptable ranges for variability. This upfront work creates the foundation for Safety, Identity, Strength, Purity, and Quality (SISPQ), the pillars of GMP compliance. Assays used to characterize potency, purity, and impurities are essential not only for defining the product, but also for linking process changes to product outcomes. When those assays fail or drift across sites, both product comparability and process understanding are undermined, eroding regulatory confidence at precisely the moment programs need reliability most.
Spending less time on characterization may save time in the short run, but it sets up years of comparability studies, regulatory setbacks, and difficult inspections later on. Investing in process discipline early allows programs to scale, sustain variability, and pass regulatory scrutiny with confidence.
Embedding the Right Expertise
Process development teams are brilliant innovators. But bringing a therapy from bench to bedside often requires voices not always embedded in those groups: experts who have taken processes from small-scale developments to commercial manufacturing lines. These professionals bring the perspective of scalability, regulatory foresight, and operational repeatability, all qualities that too often are added late, when change is costly and slow.
Working with these experts from the start helps developers avoid blind spots, adapt more smoothly to evolving regulatory requirements, and ensure that their CMC strategy is aligned with eventual commercial realities.
A Call to Elevate CMC
As regulators increase their scrutiny, the industry must evolve. In the early days, RMAT and breakthrough designations sometimes meant CMC flexibility because products were so new. Today, the industry has the expertise to do more, and the FDA expects it. Regulators now expect potency assays even in first-in-human trials, validated analytical methods, and robust comparability data for manufacturing changes.
Investors, developers, and regulators are aligned in the same mission to ensure therapies succeed and that they can be made consistently, safely, and at scale. CMC is a compliance imperative as well as a patient access issue. Therapies that stall in CMC purgatory can’t reach the patients who need them. Companies that invest early in CMC infrastructure, robust assay validation, and workforce training will not only meet FDA’s evolving standards but also differentiate commercially. That is how we move advanced therapies from promise to practice.
At Orchestra Life Sciences, we partner with innovators to align business strategy, quality systems, and manufacturing infrastructure by transforming regional momentum into real-world execution. Explore how our expert team can help position your program for success: orchestralifesciences.com.